Often researchers need to annotate multiple signatures across multiple experiments. Typically, the best way to manage this data is to process it all at once and generate a markdown report to analyze and share with collaborators. Let’s take an example where we have one experiment with many signatures.
List of 21
$ turquoise : chr [1:1902] "CLEC3A" "KCNJ3" "SCGB2A2" "SERPINA6" ...
$ blue : chr [1:1525] "GSTM1" "BMPR1B" "BMPR1B-DT" "PYDC1" ...
$ magenta : chr [1:319] "DSCAM-AS1" "VSTM2A" "UGT2B11" "CYP4Z1" ...
$ brown : chr [1:1944] "SLC25A24P1" "CPB1" "GRIA2" "CST9" ...
$ pink : chr [1:578] "MUC6" "GLRA3" "OPRPN" "ARHGAP36" ...
$ red : chr [1:681] "KCNC2" "SLC5A8" "HNRNPA1P57" "CBLN2" ...
$ darkred : chr [1:43] "OR4K12P" "GRAMD4P7" "FAR2P3" "CXADRP3" ...
$ tan : chr [1:161] "LEP" "SIK1" "TRARG1" "CIDEC" ...
$ lightcyan : chr [1:82] "CDC20B" "FOXJ1" "CDHR4" "MCIDAS" ...
$ purple : chr [1:308] "C10orf82" "GUSBP3" "IGLV10-54" "IGKV1D-13" ...
$ lightyellow : chr [1:48] "SLC6A4" "ERICH3" "GP2" "TRIM72" ...
$ cyan : chr [1:143] "NOP56P1" "FABP6" "GNAQP1" "ZNF725P" ...
$ royalblue : chr [1:47] "PCDHA12" "PCDHA11" "PCDHA4" "PCDHA1" ...
$ black : chr [1:864] "NSFP1" "USP32P2" "OCLNP1" "RN7SL314P" ...
$ yellow : chr [1:904] "NPIPB15" "MAFA-AS1" "C1orf167" "NT5CP2" ...
$ lightgreen : chr [1:60] "HIST1H2APS3" "HIST1H2AI" "HIST1H1PS1" "HIST1H3H" ...
$ darkgrey : chr [1:34] "MTND4P12" "MTRNR2L1" "MT-TT" "MTCYBP18" ...
$ darkgreen : chr [1:43] "STK19B" "SNCG" "ELANE" "TNXA" ...
$ midnightblue: chr [1:92] "LRRC26" "ARHGDIG" "TGFBR3L" "HS6ST1P1" ...
$ grey60 : chr [1:71] "KRT8P48" "KRT8P42" "KRT8P11" "CRIP1P4" ...
$ salmon : chr [1:151] "UBA52P3" "NPM1P33" "MYL6P5" "RPL29P30" ...
genesets <- hyperdb_rgsets("KEGG", "92.0")
mhyp <- hypeR(signatures, genesets, test="hypergeometric", background=36000, fdr=0.05)We have included some convenient functions that return some html for turning hyp and mhyp objects into concise tables for markdown documents. These tables can also include some plots for each individual hyp object.
rctbl_build(mhyp, show_hmaps=TRUE)Alright, how about we process all of these nested signatures and provide an example of what your markdown report might look like. Because we have a nested list of signatures, we use hyper() with the lapply function. This results in a list of mhyp objects.
lmhyp <- lapply(wgcna, hypeR, genesets, test="hypergeometric", background=36000, fdr=0.05)First start with a nice theme and include the css required to make the tables pretty.
title: "Project Title"
subtitle: "Project Subtitle"
author: "Anthony Federico"
date: "Last Modified: 'r Sys.Date()'"
output:
html_document:
theme: flatly
toc: true
toc_float: false
code_folding: hide
toc_depth: 4
css: 'r system.file("style", "rctbl.css", package="hypeR")'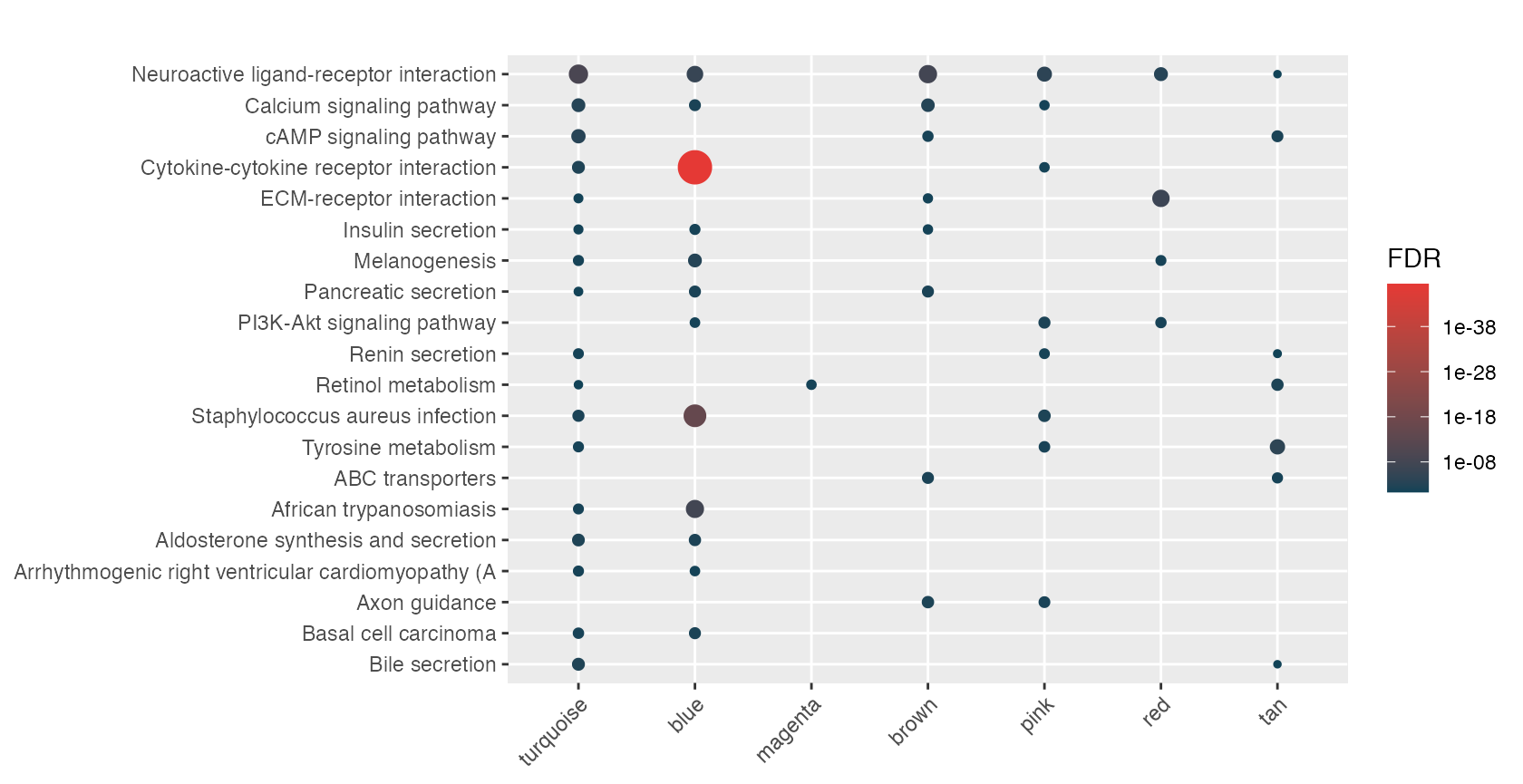
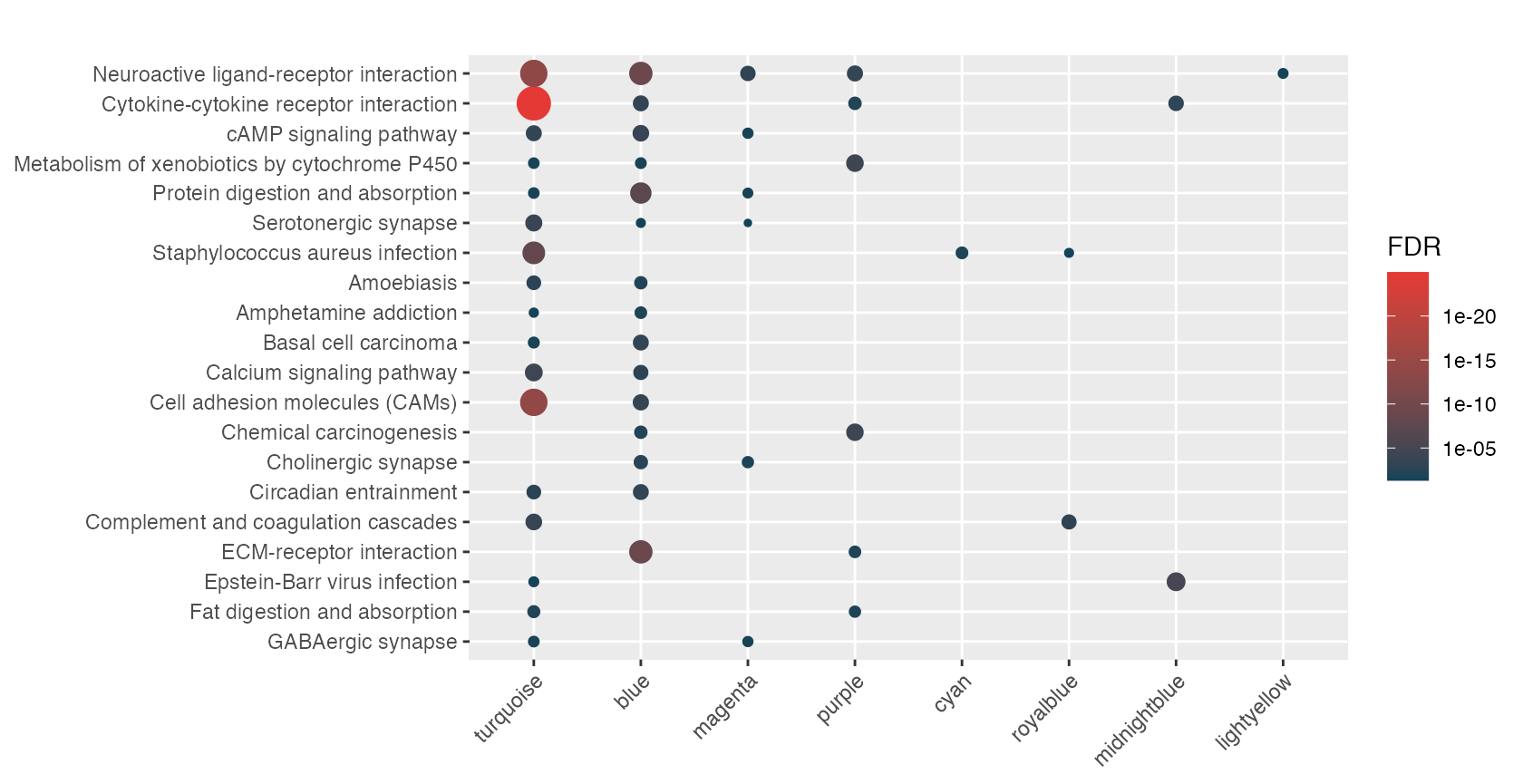
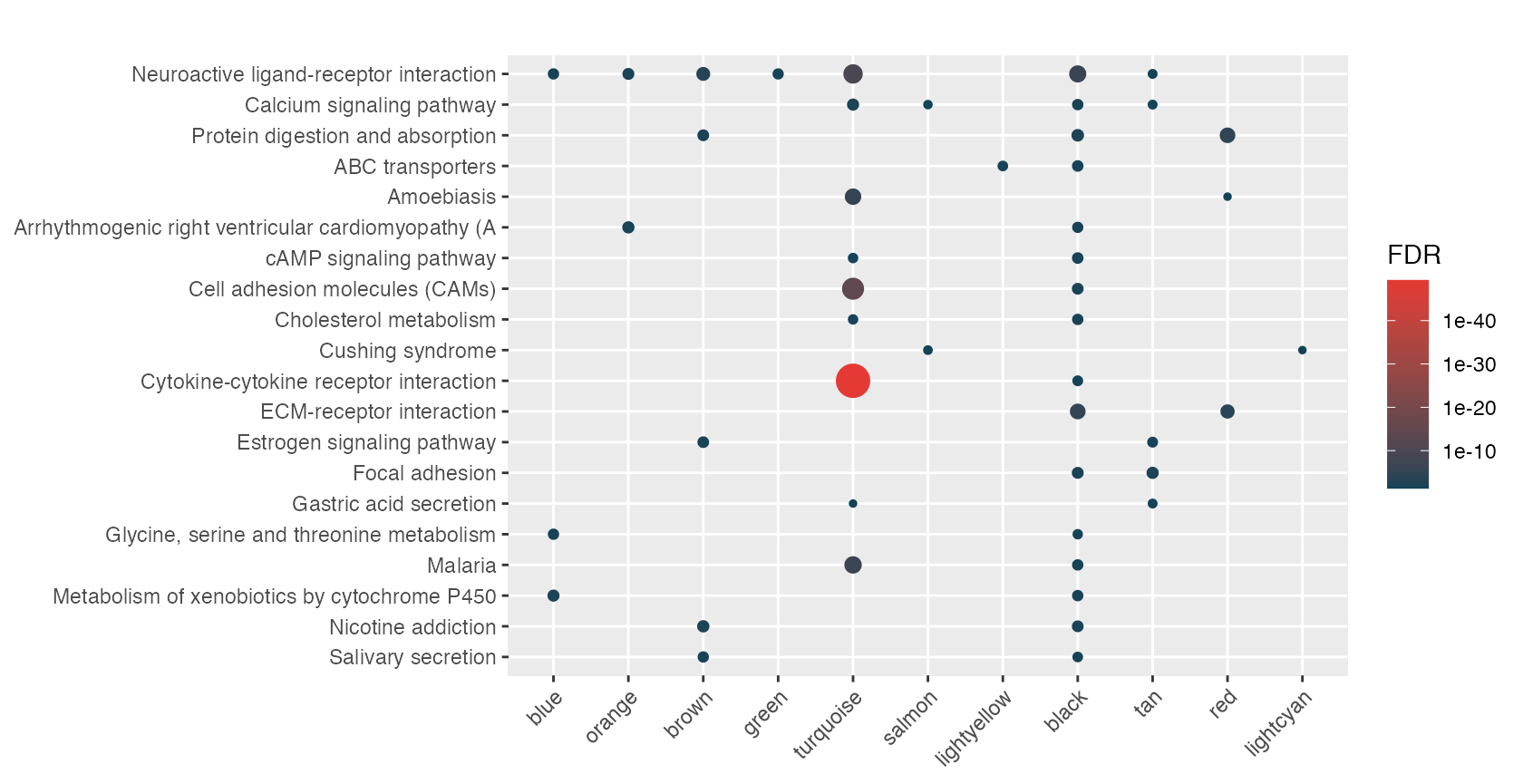
BL
hyp_dots(lmhyp$BL, merge=TRUE)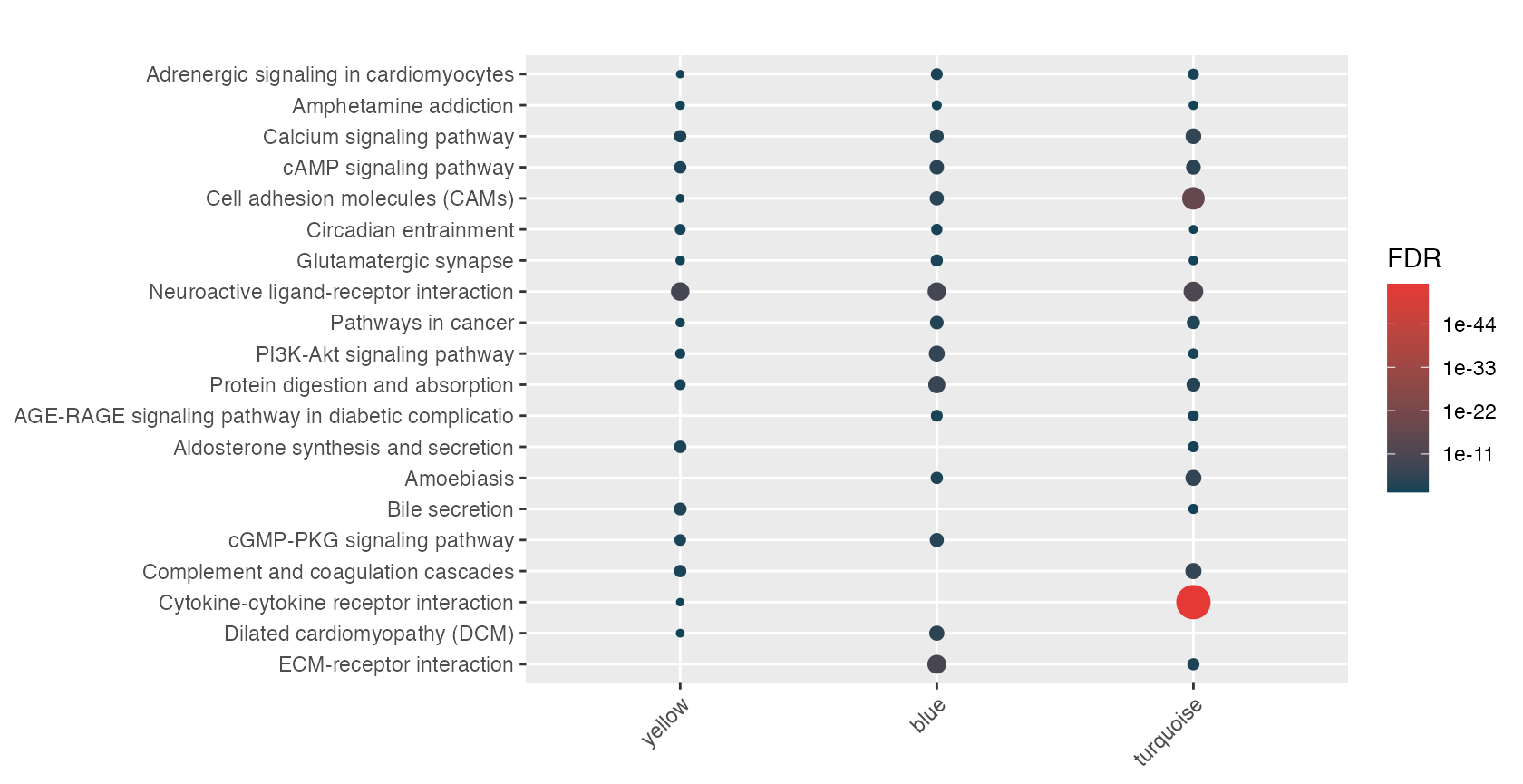
rctbl_build(lmhyp$BL, show_hmaps=TRUE)If you’re only looking at one signature you can format it with a dropdown table.
hyp <- hypeR(signatures[[1]], genesets, test="hypergeometric", background=36000, fdr=0.05)
rctbl_build(hyp)Generating Markdown Reports
For quick report generation one can use hyp_to_rmd() which will accept multiple formats, including a single hyp or multihyp object as well as a list of either, including a list of hyp or multihyp objects together. When a list of multihyp objects are passed for example, each experiment will become its own section, while each signature becomes its own tab within that section. Lists of keyword arguments can be passed for hyp_dots(), hyp_emap(), and hyp_hmap(), allowing customization of their functionality per report. See hyp_to_rmd() for details.
hyp_to_rmd(lmultihyp_obj,
file_path="hypeR.rmd",
title="hypeR Report",
subtitle="Weighted Gene Co-expression Analysis",
author="Anthony Federico, Stefano Monti",
show_dots=T,
show_emaps=T,
show_hmaps=T,
show_tables=T)