hypeR - BioC 2020 Workshop
Efficient, Scalable, and Reproducible Enrichment Workflows
Anthony Federico
Boston University School of Medicine, Boston, MAStefano Monti
Boston University School of Medicine, Boston, MAJuly 28, 2020
Source:vignettes/hyper_workshop.Rmd
hyper_workshop.RmdRequirements
We recommend the latest version of R (>= 4.0.0) but hypeR currently requires R (>= 3.6.0) to be installed directly from Github or Bioconductor.
Installation
Install the development version of the package from Github.
devtools::install_github("montilab/hypeR")
Or install the development version of the package from Bioconductor.
BiocManager::install("montilab/hypeR", version="devel")
Data
load(file.path(system.file("extdat", package="hyperworkshop"), "limma.rda")) load(file.path(system.file("extdat", package="hyperworkshop"), "wgcna.rda"))
signatures <- wgcna[[1]] str(signatures)
List of 21
$ turquoise : chr [1:1902] "CLEC3A" "KCNJ3" "SCGB2A2" "SERPINA6" ...
$ blue : chr [1:1525] "GSTM1" "BMPR1B" "BMPR1B-DT" "PYDC1" ...
$ magenta : chr [1:319] "DSCAM-AS1" "VSTM2A" "UGT2B11" "CYP4Z1" ...
$ brown : chr [1:1944] "SLC25A24P1" "CPB1" "GRIA2" "CST9" ...
$ pink : chr [1:578] "MUC6" "GLRA3" "OPRPN" "ARHGAP36" ...
$ red : chr [1:681] "KCNC2" "SLC5A8" "HNRNPA1P57" "CBLN2" ...
$ darkred : chr [1:43] "OR4K12P" "GRAMD4P7" "FAR2P3" "CXADRP3" ...
$ tan : chr [1:161] "LEP" "SIK1" "TRARG1" "CIDEC" ...
$ lightcyan : chr [1:82] "CDC20B" "FOXJ1" "CDHR4" "MCIDAS" ...
$ purple : chr [1:308] "C10orf82" "GUSBP3" "IGLV10-54" "IGKV1D-13" ...
$ lightyellow : chr [1:48] "SLC6A4" "ERICH3" "GP2" "TRIM72" ...
$ cyan : chr [1:143] "NOP56P1" "FABP6" "GNAQP1" "ZNF725P" ...
$ royalblue : chr [1:47] "PCDHA12" "PCDHA11" "PCDHA4" "PCDHA1" ...
$ black : chr [1:864] "NSFP1" "USP32P2" "OCLNP1" "RN7SL314P" ...
$ yellow : chr [1:904] "NPIPB15" "MAFA-AS1" "C1orf167" "NT5CP2" ...
$ lightgreen : chr [1:60] "HIST1H2APS3" "HIST1H2AI" "HIST1H1PS1" "HIST1H3H" ...
$ darkgrey : chr [1:34] "MTND4P12" "MTRNR2L1" "MT-TT" "MTCYBP18" ...
$ darkgreen : chr [1:43] "STK19B" "SNCG" "ELANE" "TNXA" ...
$ midnightblue: chr [1:92] "LRRC26" "ARHGDIG" "TGFBR3L" "HS6ST1P1" ...
$ grey60 : chr [1:71] "KRT8P48" "KRT8P42" "KRT8P11" "CRIP1P4" ...
$ salmon : chr [1:151] "UBA52P3" "NPM1P33" "MYL6P5" "RPL29P30" ...genesets <- msigdb_gsets("Homo sapiens", "C2", "CP:KEGG", clean=TRUE) print(genesets)
C2.CP:KEGG v7.1.1
Abc Transporters (44)
Acute Myeloid Leukemia (57)
Adherens Junction (73)
Adipocytokine Signaling Pathway (67)
Alanine Aspartate And Glutamate Metabolism (32)
Aldosterone Regulated Sodium Reabsorption (42)mhyp <- hypeR(signatures, genesets, test="hypergeometric", background=36000)
hyp_dots(mhyp, merge=TRUE, fdr=0.05, top=40, title="Co-expression Modules")
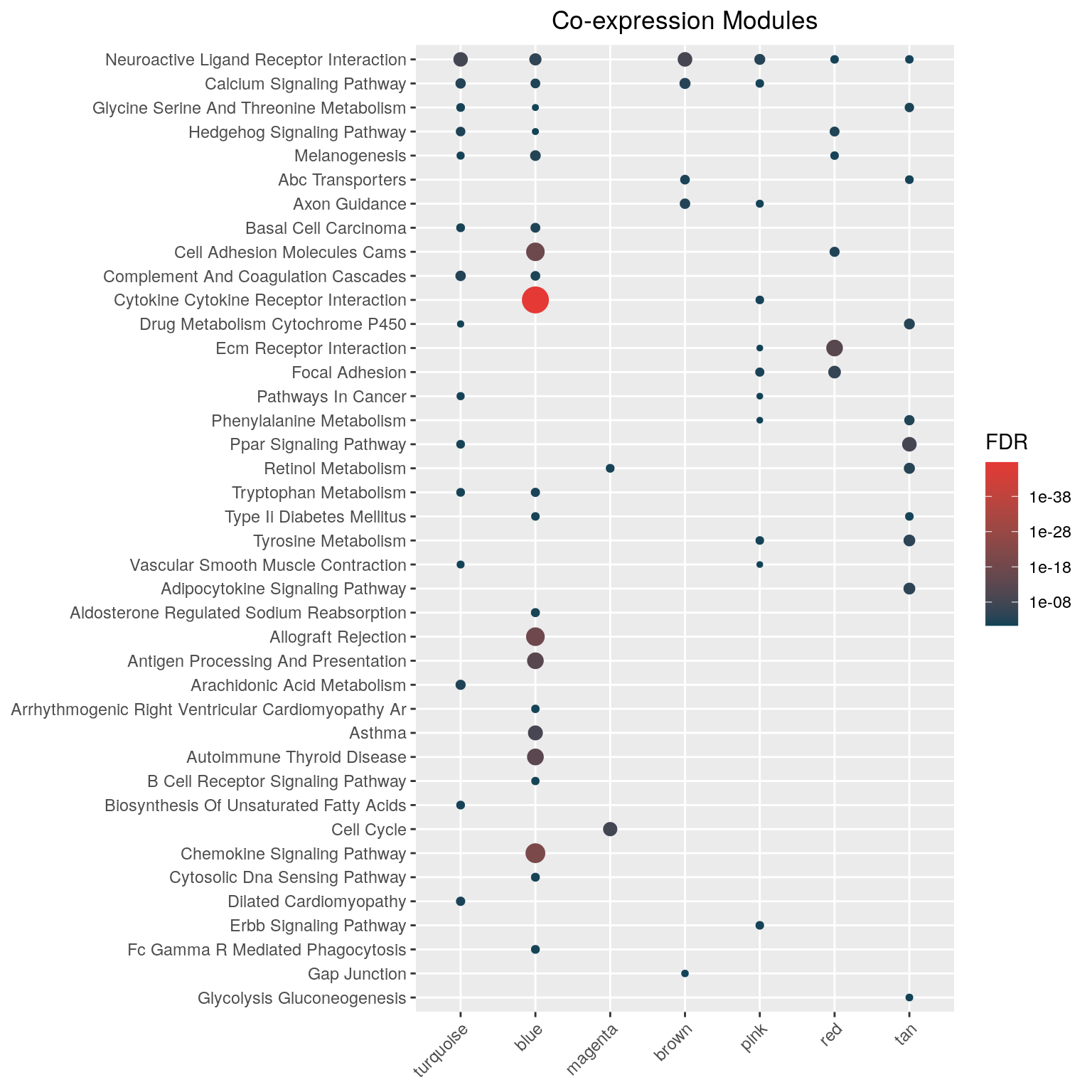
Why hypeR?
- Written purely in R
- Designed to make your life easier
- Flexible functionality and parameters
- Able to process hundreds of signatures simultaneously
- Dynamic visualizations for large amounts of data
- Highly reproducible workflows
- Report generation for sharing with collaborators
Geneset Enrichment 101
Terminology
All analyses with hypeR must include one or more signatures and genesets.
Signature
There are multiple types of enrichment analyses (e.g. hypergeometric, kstest, gsea) one can perform. Depending on the type, different kinds of signatures are expected. There are three types of signatures hypeR() expects.
# Simply a character vector of symbols (hypergeometric) signature <- c("GENE1", "GENE2", "GENE3") # A ranked character vector of symbols (kstest) ranked.signature <- c("GENE2", "GENE1", "GENE3") # A ranked named numerical vector of symbols with ranking weights (gsea) weighted.signature <- c("GENE2"=1.22, "GENE1"=0.94, "GENE3"=0.77)
Geneset
A geneset is simply a list of vectors, therefore, one can use any custom geneset in their analyses, as long as it’s appropriately defined. Additionally, hypeR() recognized object oriented genesets called gsets objects, described later.
genesets <- list("GSET1" = c("GENE1", "GENE2", "GENE3"), "GSET2" = c("GENE4", "GENE5", "GENE6"), "GSET3" = c("GENE7", "GENE8", "GENE9"))
Using a differential expression dataframe created with Limma, we will extract a signature of upregulated genes for use with a hypergeometric test and rank genes descending by their differential expression level for use with a kstest.
reactable(limma, rownames=FALSE)
We’ll also import the latest genesets from Kegg using another set of functions provided by hypeR for downloading and loading hundreds of open source genesets.
genesets <- msigdb_gsets("Homo sapiens", "C2", "CP:KEGG", clean=TRUE)
Enrichment Methods
All workflows begin with performing enrichment with hypeR(). Often we’re just interested in a single signature, as described above. In this case, hypeR() will return a hyp object. This object contains relevant information to the enrichment results, as well as plots for each geneset tested, and is recognized by downstream methods.
The most basic signature is an unranked vector of genes. This could be a differential expression signature, module of co-expressed genes, etc. As an example, we use the differential expression dataframe to filter genes that are upregulated (t > 0) and are sufficiently significant (fdr < 0.001), then extract the gene symbol column as a vector.
Unranked Signature
signature <- limma %>% dplyr::filter(t > 0 & fdr < 0.001) %>% magrittr::use_series(symbol)
length(signature)
[1] 213head(signature)
[1] "LMAN2L" "SHKBP1" "SPHK2" "AJUBA" "TJP1" "TMCC3" hyp_obj <- hypeR(signature, genesets, test="hypergeometric", background=50000, fdr=0.01, plotting=TRUE) hyp_obj$plots[[1]]
Ranked Signature
Rather than setting a specific cutoff to define a differential expression signature, one could rank genes by their expression and provide the entire ranked vector as signature. From the differential expression dataframe, we order genes descending so upregulated genes are near the top, then extract the gene symbol column as a vector.
signature <- limma %>% dplyr::arrange(desc(t)) %>% magrittr::use_series(symbol)
length(signature)
[1] 9682head(signature)
[1] "LMAN2L" "SHKBP1" "SPHK2" "AJUBA" "TJP1" "TMCC3" hyp_obj <- hypeR(signature, genesets, test="kstest", fdr=0.05, plotting=TRUE) hyp_obj$plots[[1]]
Weighted Signature
In addition to providing a ranked signature, one could also add weights by including the t-statistic of the differential expression. From the differential expression dataframe, we order genes descending so upregulated genes are near the top, then extract and deframe the gene symbol and t-statistic columns as a named vector of weights.
length(signature)
[1] 9682head(signature)
LMAN2L SHKBP1 SPHK2 AJUBA TJP1 TMCC3
6.59 6.56 6.37 6.26 6.26 6.11 hyp_obj <- hypeR(signature, genesets, test="kstest", fdr=0.05, plotting=TRUE) hyp_obj$plots[[1]]
A hyp object contains all information relevant to the enrichment analysis, including the parameters used, a dataframe of results, plots for each geneset tested, as well as the arguments used to perform the analysis. All downstream functions used for analysis, visualization, and reporting recognize hyp objects and utilize their data. Adopting an object oriented framework brings modularity to hypeR, enabling flexible and reproducible workflows.
print(hyp_obj)
(hyp)
data:
label pval fdr signature geneset overlap
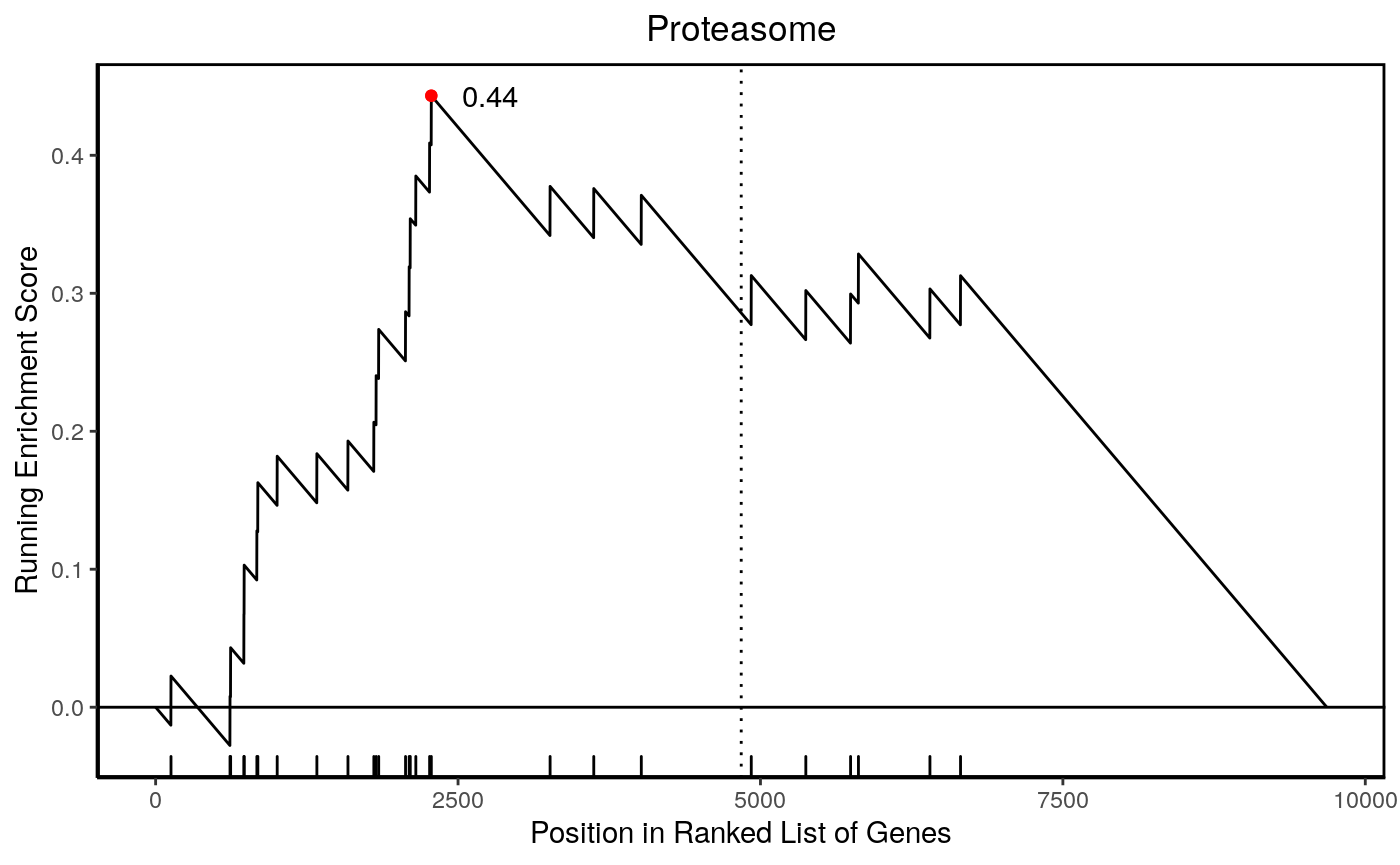
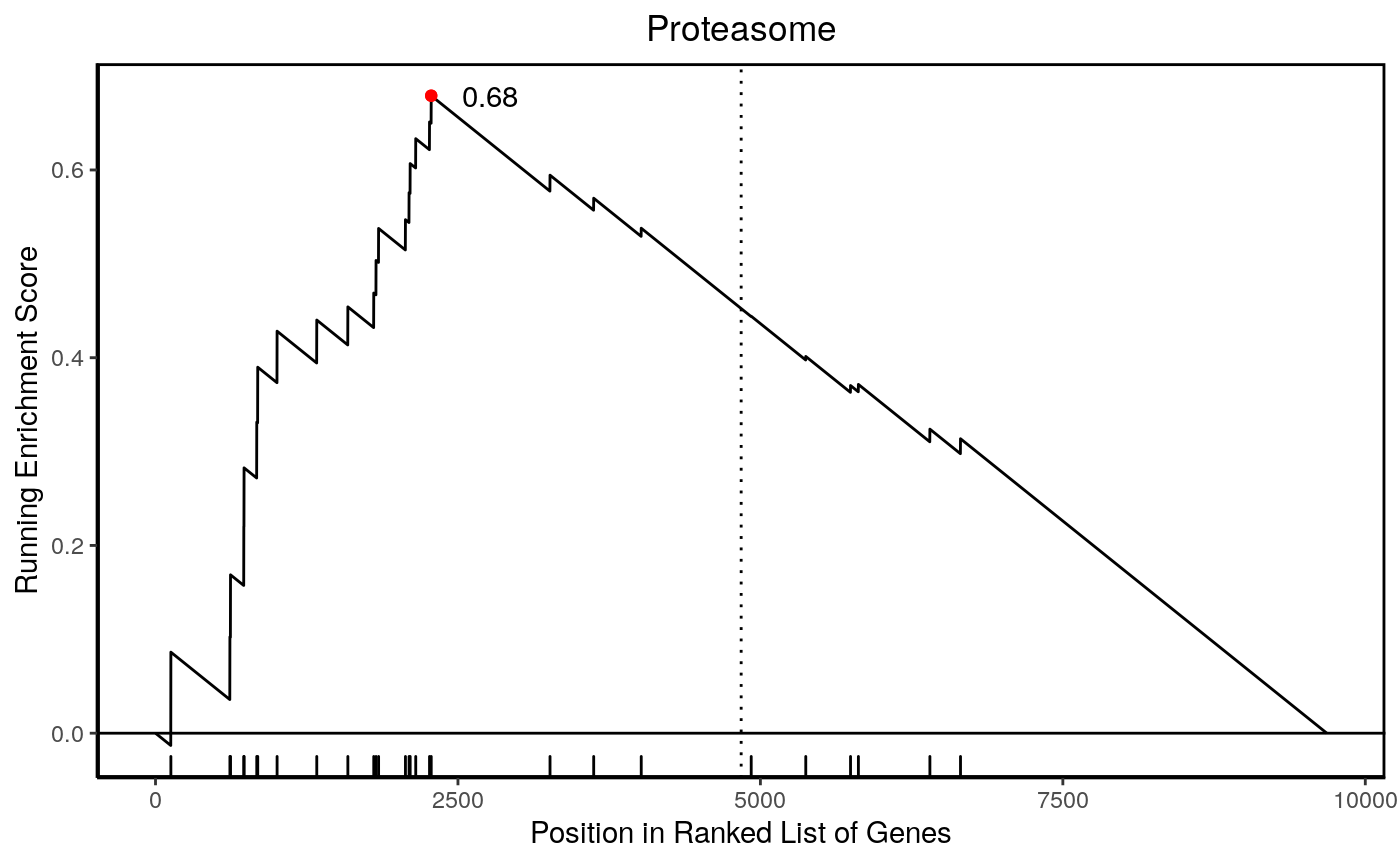
Proteasome 1.7e-05 0.0032 9682 46 28
Steroid Biosynthesis 6.4e-05 0.0060 9682 17 12
Pancreatic Cancer 1.8e-04 0.0098 9682 70 49
Neurotrophin Signaling Pathway 2.1e-04 0.0098 9682 126 87
Fc Gamma R Mediated Phagocytosis 4.8e-04 0.0180 9682 96 62
Renal Cell Carcinoma 6.6e-04 0.0200 9682 70 47
score
0.44
0.63
0.30
0.22
0.25
0.28
plots: 11 Figures
args: signature
genesets
test
background
power
absolute
pval
fdr
plotting
quiet
The hyp Methods
# Show interactive table hyp_show(hyp_obj) # Plot dots plot hyp_dots(hyp_obj) # Plot enrichment map hyp_emap(hyp_obj) # Plot hiearchy map hyp_hmap(hyp_obj) # Save to excel hyp_to_excel(hyp_obj) # Save to table hyp_to_table(hyp_obj) # Generate markdown report hyp_to_rmd(hyp_obj)
Downloading Genesets
Object Oriented Genesets
Genesets are simply a named list of character vectors which can be directly passed to hyper(). Alternatively, one can pass a gsets object, which can retain the name and version of the genesets one uses. This versioning will be included when exporting results or generating reports, which will ensure your results are reproducible.
genesets <- list("GSET1" = c("GENE1", "GENE2", "GENE3"), "GSET2" = c("GENE4", "GENE6"), "GSET3" = c("GENE7", "GENE8", "GENE9"))
Creating a gsets object is easy…
genesets <- gsets$new(genesets, name="Example Genesets", version="v1.0") print(genesets)
Example Genesets v1.0
GSET1 (3)
GSET2 (2)
GSET3 (3)And can be passed directly to hyper()…
hypeR(signature, genesets)
To aid in workflow efficiency, hypeR enables users to download genesets, wrapped as gsets objects, from multiple data sources.
Downloading msigdb Genesets
Most researchers will find the genesets hosted by msigdb are adequate to perform geneset enrichment analysis. There are various types of genesets available across multiple species.
|------------------------------------------------------------------|
| Via: R package msigdbr v7.1.1 |
|------------------------------------------------------------------|
| Available Species |
|------------------------------------------------------------------|
| Homo sapiens |
| Mus musculus |
| Drosophila melanogaster |
| Gallus gallus |
| Saccharomyces cerevisiae |
| Bos taurus |
| Caenorhabditis elegans |
| Canis lupus familiaris |
| Danio rerio |
| Rattus norvegicus |
| Sus scrofa |
|------------------------------------------------------------------|
| Available Genesets |
|------------------------------------------------------------------|
| Category Subcategory | Description |
|------------------------------------------------------------------|
| C1 | Positional |
| C2 CGP | Chemical and Genetic Perturbations |
| C2 CP | Canonical Pathways |
| C2 CP:BIOCARTA | Canonical BIOCARTA |
| C2 CP:KEGG | Canonical KEGG |
| C2 CP:PID | Canonical PID |
| C2 CP:REACTOME | Canonical REACTOME |
| C3 MIR | Motif miRNA Targets |
| C3 TFT | Motif Transcription Factor Targets |
| C4 CGN | Cancer Gene Neighborhoods |
| C4 CM | Cancer Modules |
| C5 BP | GO Biological Process |
| C5 CC | GO Cellular Component |
| C5 MF | GO Molecular Function |
| C6 | Oncogenic Signatures |
| C7 | Immunologic Signatures |
| H | Hallmark |
|------------------------------------------------------------------|
| Source: http://software.broadinstitute.org/gsea/msigdb |
|------------------------------------------------------------------|Here we download the Hallmarks genesets…
HALLMARK <- msigdb_gsets(species="Homo sapiens", category="H") print(HALLMARK)
H v7.1.1
HALLMARK_ADIPOGENESIS (200)
HALLMARK_ALLOGRAFT_REJECTION (200)
HALLMARK_ANDROGEN_RESPONSE (100)
HALLMARK_ANGIOGENESIS (36)
HALLMARK_APICAL_JUNCTION (200)
HALLMARK_APICAL_SURFACE (44)We can also clean them up by removing the first leading common substring…
HALLMARK <- msigdb_gsets(species="Homo sapiens", category="H", clean=TRUE) print(HALLMARK)
H v7.1.1
Adipogenesis (200)
Allograft Rejection (200)
Androgen Response (100)
Angiogenesis (36)
Apical Junction (200)
Apical Surface (44)This can be passed directly to hypeR()…
hypeR(signature, genesets=HALLMARK)
Other commonly used genesets include Biocarta, Kegg, and Reactome…
BIOCARTA <- msigdb_gsets(species="Homo sapiens", category="C2", subcategory="CP:BIOCARTA") KEGG <- msigdb_gsets(species="Homo sapiens", category="C2", subcategory="CP:KEGG") REACTOME <- msigdb_gsets(species="Homo sapiens", category="C2", subcategory="CP:REACTOME")
Downloading enrichr Genesets
If msigdb genesets are not sufficient, we have also provided another set of functions for downloading and loading other publicly available genesets. This is facilitated by interfacing with the publicly available libraries hosted by enrichr.
available <- enrichr_available() reactable(available)
KEGG <- enrichr_gsets("KEGG_2019_Human") print(KEGG)
KEGG_2019_Human (Enrichr) Downloaded: 2020-07-28
ABC transporters (45)
AGE-RAGE signaling pathway in diabetic complications (100)
AMPK signaling pathway (120)
Acute myeloid leukemia (66)
Adherens junction (72)
Adipocytokine signaling pathway (69)Note: These libraries do not have a systematic versioning scheme, however the date downloaded will be recorded.
Additionally download other species if you aren’t working with human or mouse genes!
yeast <- enrichr_gsets("GO_Biological_Process_2018", db="YeastEnrichr") worm <- enrichr_gsets("GO_Biological_Process_2018", db="WormEnrichr") fish <- enrichr_gsets("GO_Biological_Process_2018", db="FishEnrichr") fly <- enrichr_gsets("GO_Biological_Process_2018", db="FlyEnrichr")
Visualization
signature <- limma %>% dplyr::arrange(desc(t)) %>% magrittr::use_series(symbol) head(signature)
[1] "LMAN2L" "SHKBP1" "SPHK2" "AJUBA" "TJP1" "TMCC3" genesets <- msigdb_gsets("Homo sapiens", "C2", "CP:REACTOME", clean=TRUE) hyp_obj <- hypeR(signature, genesets, test="kstest", fdr=0.01)
To visualize the results, just pass the hyp object to any downstream functions.
Dots Plot
One can visualize the top enriched genesets using hyp_dots() which returns a horizontal dots plot. Each dot is a geneset, where the color represents the significance and the size signifies the geneset size.
hyp_dots(hyp_obj)
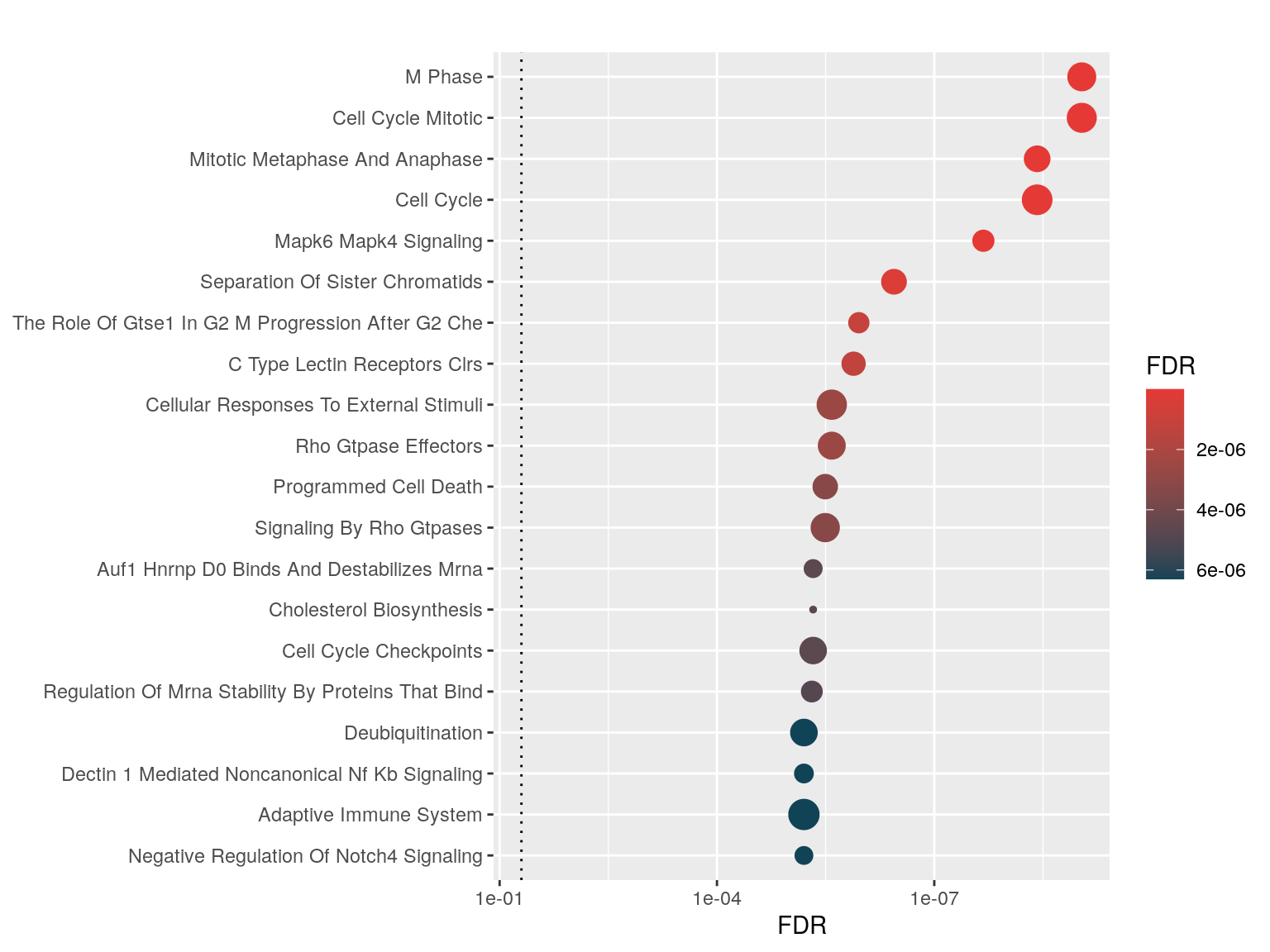
Enrichment Map
One can visualize the top enriched genesets using hyp_emap() which will return an enrichment map. Each node represents a geneset, where the shade of red indicates the normalized significance of enrichment. Hover over the node to view the raw value. Edges represent geneset similarity, calculated by either jaccard or overlap similarity metrics.
hyp_emap(hyp_obj, similarity_cutoff=0.70)
Hiearchy Map
When dealing with hundreds of genesets, it’s often useful to understand the relationships between them. This allows researchers to summarize many enriched pathways as more general biological processes. To do this, we rely on curated relationships defined between them.
Relational Genesets
For example, Reactome conveniently defines their genesets in a hiearchy of pathways. This data can be formatted into a relational genesets object called rgsets.
genesets <- hyperdb_rgsets("REACTOME", version="70.0")
Relational genesets have three data atrributes including gsets, nodes, and edges. The genesets attribute includes the geneset information for the leaf nodes of the hiearchy, the nodes attribute describes all nodes in the hierarchy, including internal nodes, and the edges attribute describes the edges in the hiearchy.
print(genesets)
REACTOME v70.0
Genesets
2-LTR circle formation (13)
5-Phosphoribose 1-diphosphate biosynthesis (3)
A tetrasaccharide linker sequence is required for GAG synthesis (26)
ABC transporters in lipid homeostasis (18)
ABO blood group biosynthesis (3)
ADP signalling through P2Y purinoceptor 1 (25)
Nodes
label
R-HSA-164843 2-LTR circle formation
R-HSA-73843 5-Phosphoribose 1-diphosphate biosynthesis
R-HSA-1971475 A tetrasaccharide linker sequence is required for GAG synthesis
R-HSA-5619084 ABC transporter disorders
R-HSA-1369062 ABC transporters in lipid homeostasis
R-HSA-382556 ABC-family proteins mediated transport
id length
R-HSA-164843 R-HSA-164843 13
R-HSA-73843 R-HSA-73843 3
R-HSA-1971475 R-HSA-1971475 26
R-HSA-5619084 R-HSA-5619084 77
R-HSA-1369062 R-HSA-1369062 18
R-HSA-382556 R-HSA-382556 22
Edges
from to
1 R-HSA-109581 R-HSA-109606
2 R-HSA-109581 R-HSA-169911
3 R-HSA-109581 R-HSA-5357769
4 R-HSA-109581 R-HSA-75153
5 R-HSA-109582 R-HSA-140877
6 R-HSA-109582 R-HSA-202733Passing relational genesets works natively with hypeR().
hyp_obj <- hypeR(signature, genesets, test="kstest", fdr=0.01)
One can visualize the top enriched genesets using hyp_hmap() which will return a hiearchy map. Each node represents a geneset, where the shade of the gold border indicates the normalized significance of enrichment. Hover over the leaf nodes to view the raw value. Double click internal nodes to cluster their first degree connections. Edges represent a directed relationship between genesets in the hiearchy. Note: This function only works when the hyp object was inialized with an rgsets object.
hyp_hmap(hyp_obj, top=30)
Use Case: Gene Co-Expression Analysis
Often computational biologists must process, interpret, and share large amounts of biological data. A common example is interpreting gene co-expression modules across one or more phenotype, resulting in potentially hundreds of signatures to annotate. Here we have signatures of gene co-expression modules, generated with WGCNA, for multiple breast cancer subtypes
Problem: Annotate gene co-expression modules across one or more phenotype, resulting in potentially hundreds of signatures to annotate. ## Example Data
We’re using publicly available RNA-seq data from breast cancer samples from The Cancer Genome Atlas. The data has been normalized with DESeq2, log-transformed, and subtyped using PAM50 signatures.
Weighted Gene Co-Expression Analysis (WGCNA)
run.wgcna(brca)
Apply to BRCA Subtypes
Isolate tumor samples with subtypes LumA, LumB, Her2, and Basal.
subtypes <- c("LA", "LB", "H2", "BL") brca.tumors <- brca[, brca$subtype %in% subtypes] table(brca.tumors$subtype)
wgcna <- list("LA" = run.wgcna(brca.tumors, subtype="LA"), "LB" = run.wgcna(brca.tumors, subtype="LB"), "H2" = run.wgcna(brca.tumors, subtype="H2"), "BL" = run.wgcna(brca.tumors, subtype="BL"))
Resulting 87 Signatures to Annotate…
str(wgcna)
List of 4
$ LA:List of 21
..$ turquoise : chr [1:1902] "CLEC3A" "KCNJ3" "SCGB2A2" "SERPINA6" ...
..$ blue : chr [1:1525] "GSTM1" "BMPR1B" "BMPR1B-DT" "PYDC1" ...
..$ magenta : chr [1:319] "DSCAM-AS1" "VSTM2A" "UGT2B11" "CYP4Z1" ...
..$ brown : chr [1:1944] "SLC25A24P1" "CPB1" "GRIA2" "CST9" ...
..$ pink : chr [1:578] "MUC6" "GLRA3" "OPRPN" "ARHGAP36" ...
..$ red : chr [1:681] "KCNC2" "SLC5A8" "HNRNPA1P57" "CBLN2" ...
..$ darkred : chr [1:43] "OR4K12P" "GRAMD4P7" "FAR2P3" "CXADRP3" ...
..$ tan : chr [1:161] "LEP" "SIK1" "TRARG1" "CIDEC" ...
..$ lightcyan : chr [1:82] "CDC20B" "FOXJ1" "CDHR4" "MCIDAS" ...
..$ purple : chr [1:308] "C10orf82" "GUSBP3" "IGLV10-54" "IGKV1D-13" ...
..$ lightyellow : chr [1:48] "SLC6A4" "ERICH3" "GP2" "TRIM72" ...
..$ cyan : chr [1:143] "NOP56P1" "FABP6" "GNAQP1" "ZNF725P" ...
..$ royalblue : chr [1:47] "PCDHA12" "PCDHA11" "PCDHA4" "PCDHA1" ...
..$ black : chr [1:864] "NSFP1" "USP32P2" "OCLNP1" "RN7SL314P" ...
..$ yellow : chr [1:904] "NPIPB15" "MAFA-AS1" "C1orf167" "NT5CP2" ...
..$ lightgreen : chr [1:60] "HIST1H2APS3" "HIST1H2AI" "HIST1H1PS1" "HIST1H3H" ...
..$ darkgrey : chr [1:34] "MTND4P12" "MTRNR2L1" "MT-TT" "MTCYBP18" ...
..$ darkgreen : chr [1:43] "STK19B" "SNCG" "ELANE" "TNXA" ...
..$ midnightblue: chr [1:92] "LRRC26" "ARHGDIG" "TGFBR3L" "HS6ST1P1" ...
..$ grey60 : chr [1:71] "KRT8P48" "KRT8P42" "KRT8P11" "CRIP1P4" ...
..$ salmon : chr [1:151] "UBA52P3" "NPM1P33" "MYL6P5" "RPL29P30" ...
$ LB:List of 22
..$ turquoise : chr [1:5327] "CLEC3A" "PSPHP1" "PRAME" "CPB1" ...
..$ black : chr [1:195] "TFAP2B" "MUC5B" "ZIC2" "CRABP1" ...
..$ blue : chr [1:2282] "SCGB2A2" "KCNJ3" "ASCL1" "CGA" ...
..$ magenta : chr [1:161] "BEX1" "ONECUT2" "SULT4A1" "PCSK1N" ...
..$ red : chr [1:199] "PVALB" "PIP" "SERPINA6" "COL2A1" ...
..$ purple : chr [1:151] "ADIPOQ" "TRARG1" "ADH1B" "CIDEC" ...
..$ cyan : chr [1:70] "KRT14" "KRT6B" "KRT5" "KLK5" ...
..$ salmon : chr [1:85] "PCDHA11" "PCDHA12" "DIO1" "PCDHA4" ...
..$ lightcyan : chr [1:59] "CDC20B" "FOXJ1" "MCIDAS" "CCNO" ...
..$ yellow : chr [1:244] "TPSG1" "PRTN3" "PYY" "AZU1" ...
..$ royalblue : chr [1:46] "DLGAP1" "OR7E22P" "PTPRN2" "DCX" ...
..$ lightgreen : chr [1:56] "HIST1H3G" "HIST1H2APS3" "HIST1H1D" "HIST1H2AI" ...
..$ midnightblue: chr [1:68] "B3GALT5" "PCP4" "B3GALT5-AS1" "IFI27" ...
..$ pink : chr [1:166] "PNMT" "TBX1" "MAPK15" "KRT13" ...
..$ lightyellow : chr [1:49] "LINC02593" "SAMD11" "EFNA2" "ARHGDIG" ...
..$ darkgreen : chr [1:31] "DOK7" "ITPKA" "CACNA1F" "NHLRC4" ...
..$ green : chr [1:230] "GAPDHP1" "RPS14P8" "MT-TV" "GAPDHP35" ...
..$ greenyellow : chr [1:122] "FAM169A" "MIR4768" "ERVE-1" "RPS23P6" ...
..$ brown : chr [1:273] "PWAR5" "SPATA46" "MTND1P8" "EML5" ...
..$ tan : chr [1:88] "GCGR" "TRIM54" "CYP2E1" "GOLGA8A" ...
..$ grey60 : chr [1:57] "KRT8P42" "KRT8P48" "KRT8P11" "KRT8P3" ...
..$ darkred : chr [1:41] "ATP1A2" "PCDH19" "ADRA1A" "FRAS1" ...
$ H2:List of 25
..$ blue : chr [1:1940] "CLEC3A" "S100A7" "CYP2B7P" "CASP14" ...
..$ orange : chr [1:631] "GSTM1" "RPSAP53" "LINC01833" "LINC00461" ...
..$ brown : chr [1:885] "SLC5A8" "DSCAM-AS1" "PRAME" "KCNJ3" ...
..$ darkred : chr [1:54] "NXPH1" "PNMT" "SCGN" "GACAT3" ...
..$ green : chr [1:586] "ARHGAP40" "CYP4F8" "GATD3A" "IRX4" ...
..$ darkgreen : chr [1:49] "MUCL1" "BRINP3" "C2CD4A" "DMRTC2" ...
..$ turquoise : chr [1:2389] "MKRN3" "MUC6" "SOX2" "GUSBP3" ...
..$ cyan : chr [1:137] "HMGCS2" "ONECUT2" "PLAC9P1" "SLC7A4" ...
..$ salmon : chr [1:178] "PYDC1" "PCDHB2" "LINC01342" "ARHGDIG" ...
..$ lightyellow : chr [1:65] "ABCC12" "LRRC31" "MUC19" "CEACAM5" ...
..$ black : chr [1:554] "TRARG1" "ADIPOQ" "CIDEA" "CIDEC" ...
..$ tan : chr [1:201] "OLFM4" "TCN1" "KLK11" "CALML3" ...
..$ red : chr [1:578] "PCDHA11" "PCDHA12" "PCDHA4" "USP32P1" ...
..$ darkgrey : chr [1:36] "FOXJ1" "CDC20B" "LRRIQ1" "CCNO" ...
..$ royalblue : chr [1:60] "HIST1H3G" "HIST1H2APS3" "HIST1H2APS4" "HIST1H1D" ...
..$ lightcyan : chr [1:118] "PCSK1N" "ZNF725P" "NCAM2" "RP1" ...
..$ grey60 : chr [1:90] "LRRC26" "AMH" "TGFBR3L" "LKAAEAR1" ...
..$ magenta : chr [1:357] "ZSCAN1" "HDGFP1" "PRSS29P" "LINC00930" ...
..$ darkturquoise: chr [1:49] "DYDC2" "ATP6V1B1" "PRSS50" "PRSS45" ...
..$ pink : chr [1:453] "SPATA46" "ZNF727" "XIST" "ZSCAN23" ...
..$ midnightblue : chr [1:127] "ADGRV1" "LINC02211" "LRRC37A9P" "CCDC144B" ...
..$ lightgreen : chr [1:90] "FAM90A2P" "VN1R5" "FAM71F1" "NXF5" ...
..$ white : chr [1:32] "ELANE" "CFD" "C2CD4C" "GSC" ...
..$ darkorange : chr [1:33] "ITPKA" "C20orf204" "STXBP5L" "SULT2B1" ...
..$ purple : chr [1:308] "MTND4P24" "MTRNR2L1" "GAPDHP68" "RPL7P16" ...
$ BL:List of 19
..$ yellow : chr [1:801] "ADIPOQ" "ADH1B" "GABRA5" "LINP1" ...
..$ pink : chr [1:128] "KRT81" "LINC00518" "VXN" "MUC15" ...
..$ purple : chr [1:75] "S100A7" "CASP14" "KRT6C" "TMPRSS4" ...
..$ blue : chr [1:2280] "CDH19" "LINC01667" "LINC01224" "HILS1" ...
..$ turquoise : chr [1:4149] "FDCSP" "HOXC12" "COL2A1" "AFAP1-AS1" ...
..$ grey60 : chr [1:53] "DSG3" "KLK11" "SCEL" "KLK7" ...
..$ brown : chr [1:988] "PEG3" "KLHL34" "SERPINB4" "ZIM2-AS1" ...
..$ cyan : chr [1:63] "KIF1A" "PRDM13" "LINC01606" "TCEAL2" ...
..$ lightcyan : chr [1:57] "CXCL17" "ODAM" "BMP5" "ART3" ...
..$ tan : chr [1:67] "SLC6A15" "ALX1" "KCNV1" "MYO3A" ...
..$ green : chr [1:427] "CALML5" "GSTM1" "PRAC2" "PSPHP1" ...
..$ magenta : chr [1:126] "WIF1" "PRSS33" "COL9A1" "SCRG1" ...
..$ red : chr [1:277] "MINOS1P3" "COX6CP1" "RPL10P6" "ZBTB8OSP2" ...
..$ lightyellow: chr [1:46] "IRX4" "RSPO4" "LHX2" "MGAT5B" ...
..$ greenyellow: chr [1:69] "IVL" "KRT6A" "LY6D" "KRT4" ...
..$ salmon : chr [1:66] "CEACAM6" "SLURP1" "S100P" "LCN2" ...
..$ royalblue : chr [1:36] "MTND4P24" "MTND1P23" "MTATP8P2" "MT-TT" ...
..$ black : chr [1:246] "GAPDHP1" "GAPDHP35" "RPS14P8" "RPL26P37" ...
..$ lightgreen : chr [1:46] "HIST1H2APS3" "HIST1H3C" "HIST1H3A" "HIST1H2AE" ...Processing Multiple Signatures
We can feed hypeR() one or more signatures. In the case of multiple signatures, a multihyp object will be returned. This object is essentially just multiple hyp objects. However it is recognized and handled differently by downstream methods. Lets start by annotating signatures for the first subtype.
genesets <- msigdb_gsets(species="Homo sapiens", category="C2", subcategory="CP:REACTOME", clean=TRUE) mhyp <- hypeR(wgcna[["LA"]], genesets=genesets, background=36812, fdr=0.25)
print(mhyp)
(multihyp)
(hyp) turquoise
118 x 8
(hyp) blue
121 x 8
(hyp) magenta
67 x 8
(hyp) brown
59 x 8
(hyp) pink
94 x 8
(hyp) red
97 x 8
(hyp) darkred
0 x 8
(hyp) tan
62 x 8
(hyp) lightcyan
0 x 8
(hyp) purple
28 x 8
(hyp) lightyellow
0 x 8
(hyp) cyan
0 x 8
(hyp) royalblue
0 x 8
(hyp) black
0 x 8
(hyp) yellow
5 x 8
(hyp) lightgreen
0 x 8
(hyp) darkgrey
0 x 8
(hyp) darkgreen
0 x 8
(hyp) midnightblue
0 x 8
(hyp) grey60
0 x 8
(hyp) salmon
0 x 8Alternatively, we can pass a background distribution of genes, which will reduce genesets only to the genes used to generate the signatures.
Let’s see the overall enrichment across signatures…
hyp_dots(mhyp, abrv=100, merge=TRUE)
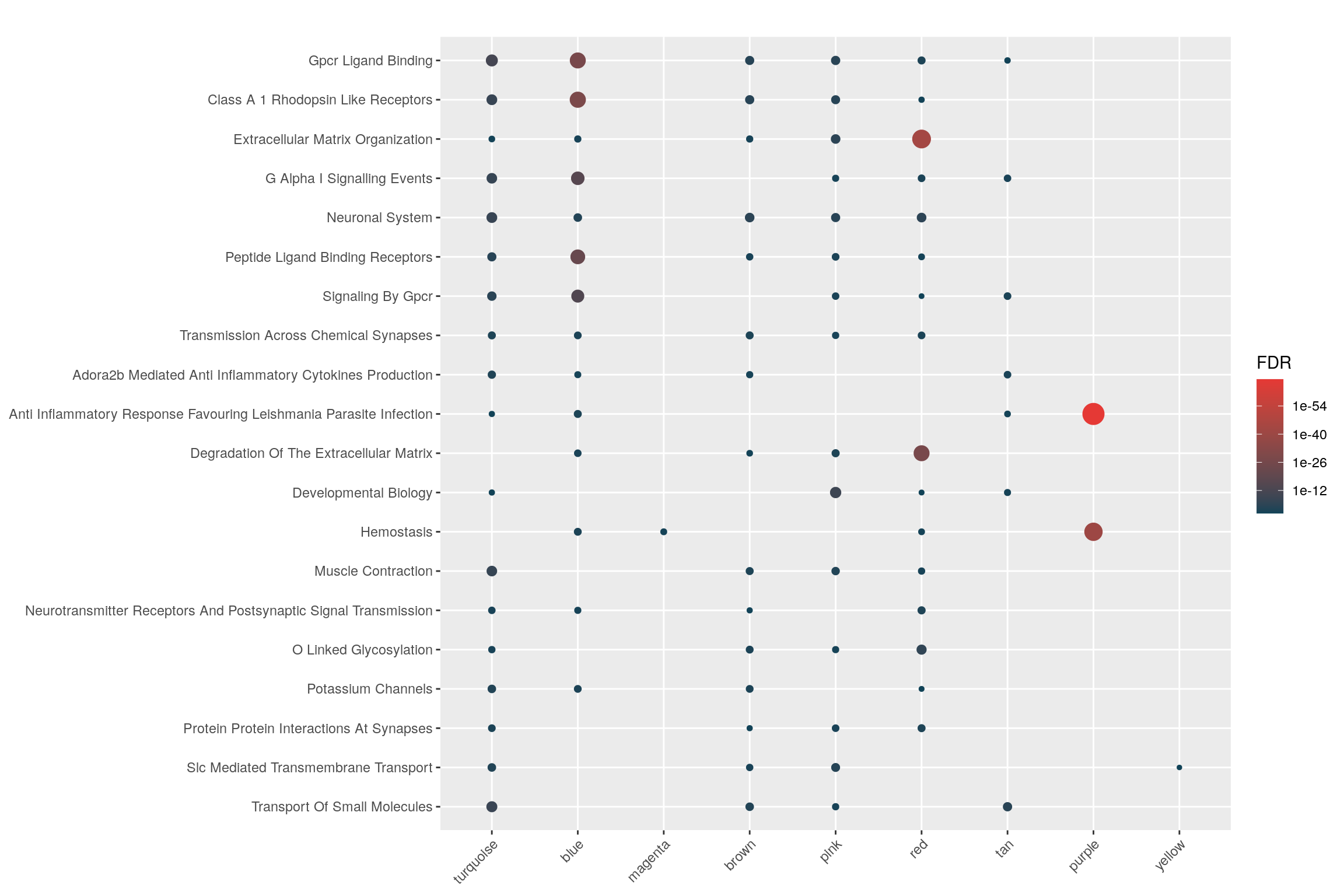
Exporting Results
With hyp_to_table each signature is exported as its own table in a single directory…
hyp_to_table(mhyp, file_path="hypeR")
With hyp_to_excel each signature is exported to its own sheet…
hyp_to_excel(mhyp, file_path="hypeR.xlsx")
A Note on Reproducibility
When data is exported, hypeR makes sure to include all parameters and versioning required for reproducibility.
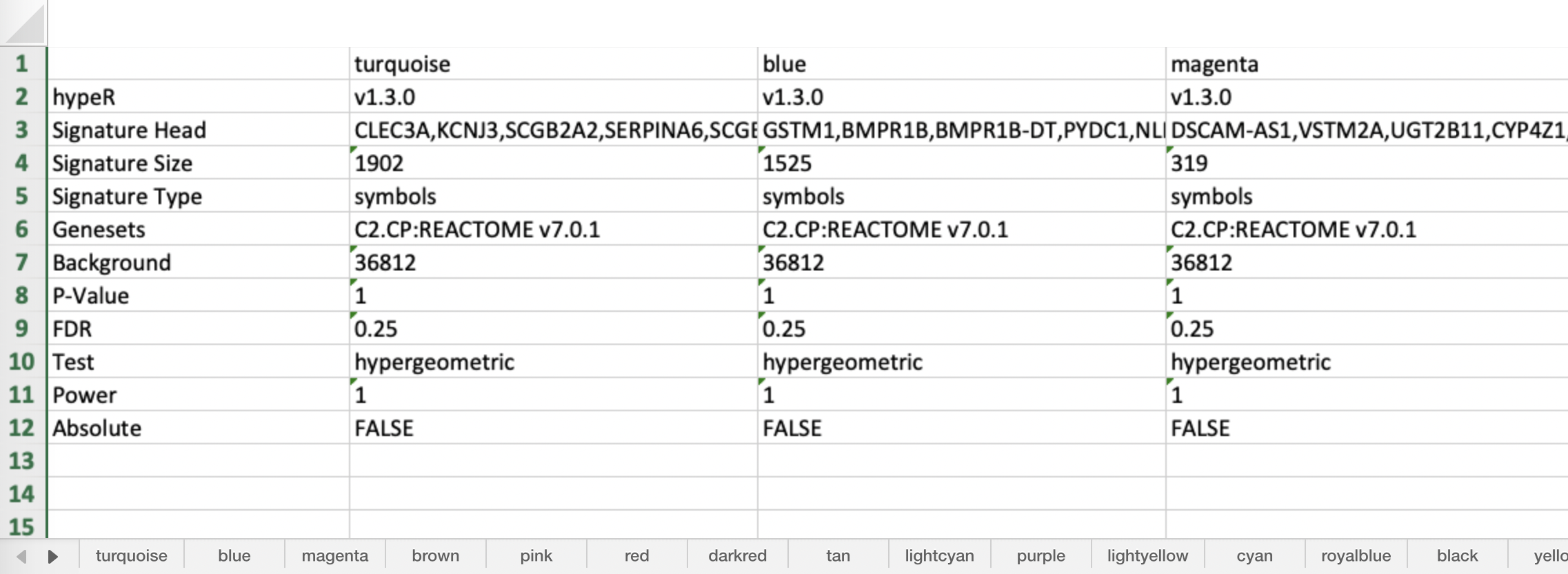
In the case of exporting tables, reproducibility information is in the header of each signature…
#hypeR v1.3.0 #Signature Head CLEC3A,KCNJ3,SCGB2A2,SERPINA6,SCGB1D2,RPSAP53 #Signature Size 1902 #Signature Type symbols #Genesets C2.CP:REACTOME v7.0.1 #Background 36812 #P-Value 1 #FDR 0.25 #Test hypergeometric #Power 1 #Absolute FALSE
For excel sheets, there is a dedicated tab with reproducibility information for all signatures…
Generating Reports
Often researchers need to annotate multiple signatures across multiple experiments. Typically, the best way to manage this data is to process it all at once and generate a markdown report to analyze and share with collaborators. Let’s take an example where we have one experiment with many signatures.
In the current example, we want to annotate dozens of co-expression modules for multiple breast cancer subtypes. We have included some convenient functions that return some html for turning hyp and mhyp objects into concise tables for markdown documents. These tables can also include some plots for each individual hyp object.
rctbl_mhyp(mhyp)
Alright, how about we process all of these nested signatures and provide an example of what your markdown report might look like. Because we have a nested list of signatures, we use hyper() with the lapply function. This results in a list of mhyp objects.
genesets <- hyperdb_rgsets("KEGG", "92.0") lmhyp <- lapply(wgcna, hypeR, genesets, test="hypergeometric", background=36000, fdr=0.05)
First start with a nice theme and include the css required to make the tables pretty.
title: "Project Title" subtitle: "Project Subtitle" author: "Anthony Federico" date: "Last Modified: 'r Sys.Date()'" output: html_document: theme: flatly toc: true toc_float: false code_folding: hide toc_depth: 4 css: 'r system.file("style", "rctbl.css", package="hypeR")'
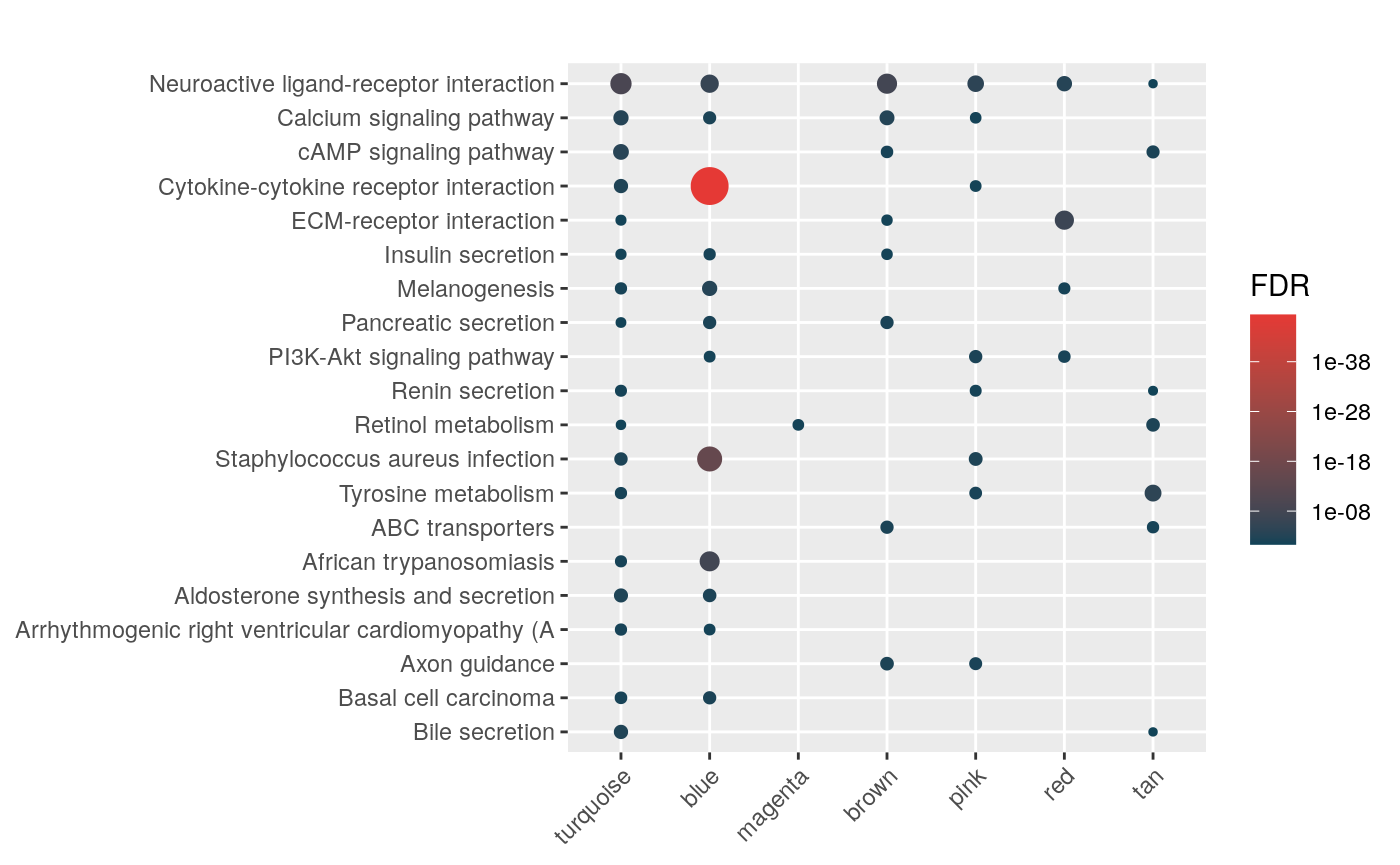
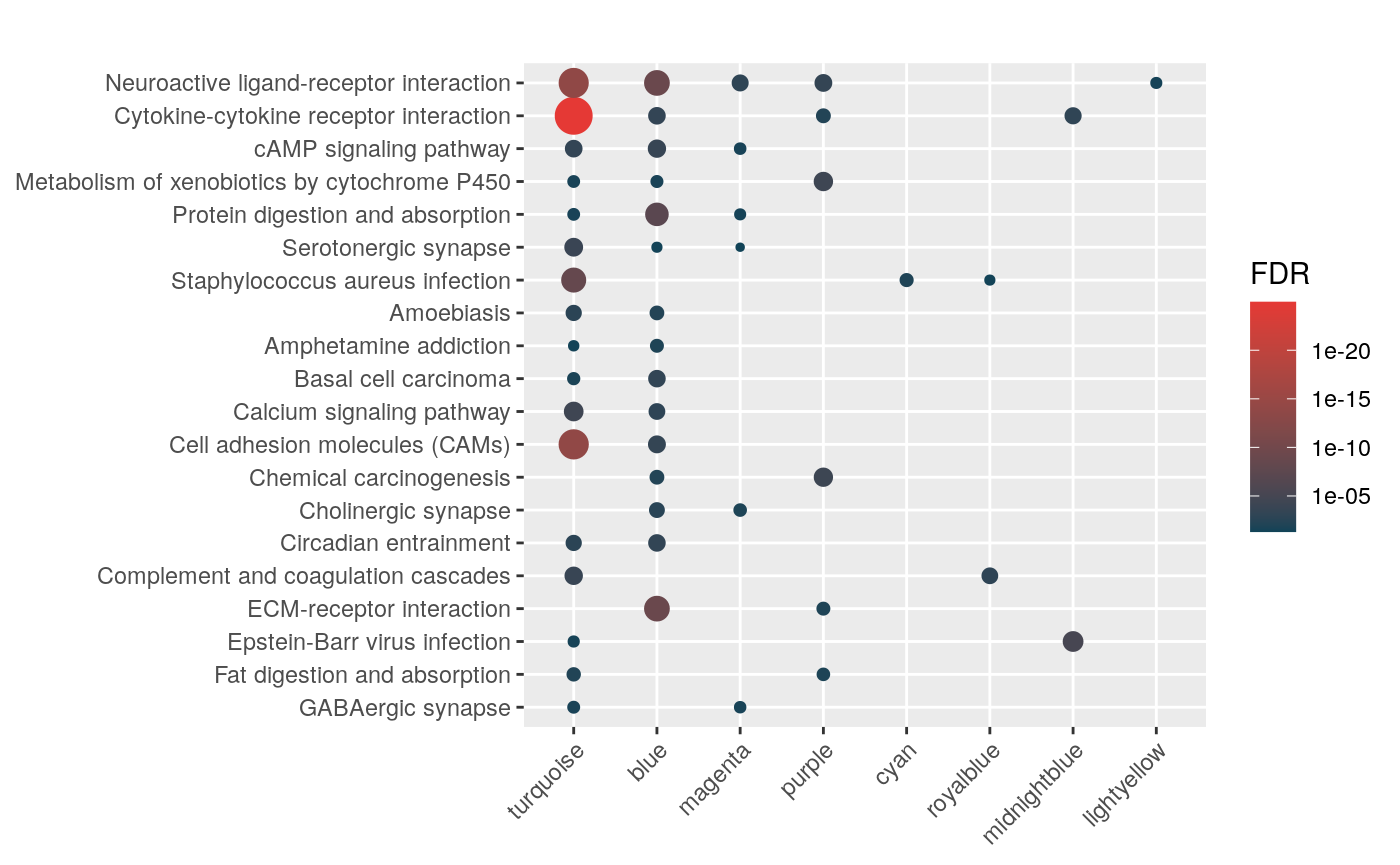
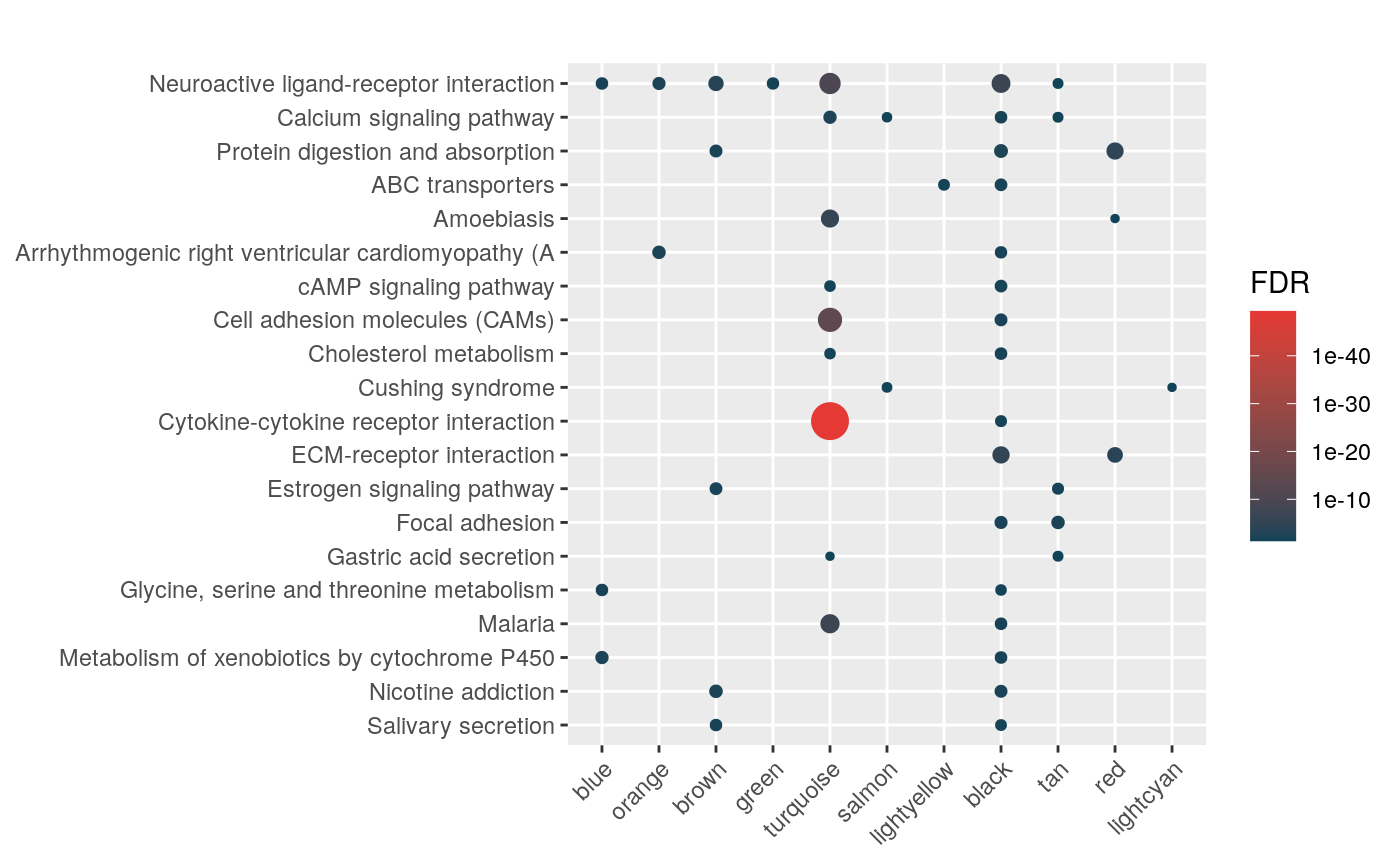
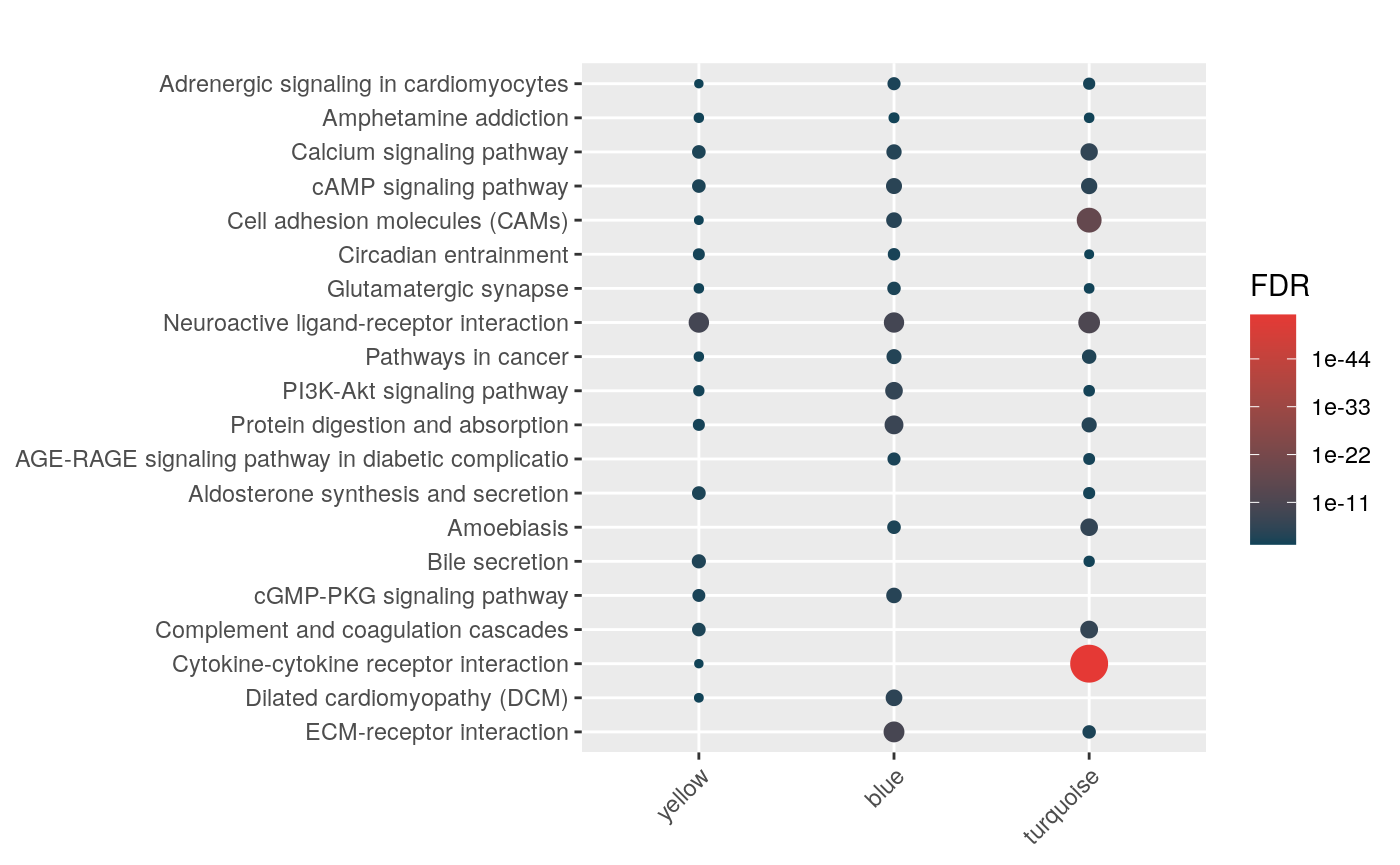
BL
hyp_dots(lmhyp$BL, merge=TRUE)
rctbl_mhyp(lmhyp$BL, show_hmaps=TRUE)
For quick report generation one can use hyp_to_rmd() which will accept multiple formats, including a single hyp or multihyp object as well as a list of either, including a list of hyp or multihyp objects together. When a list of multihyp objects are passed for example, each experiment will become its own section, while each signature becomes its own tab within that section. Lists of keyword arguments can be passed for hyp_dots(), hyp_emap(), and hyp_hmap(), allowing customization of their functionality per report. See hyp_to_rmd() for details.
hyp_to_rmd(lmultihyp_obj, file_path="hypeR.rmd", title="hypeR Report", subtitle="Weighted Gene Co-expression Analysis", author="Anthony Federico, Stefano Monti", show_dots=T, show_emaps=T, show_hmaps=T, show_tables=T)
Shiny Modules
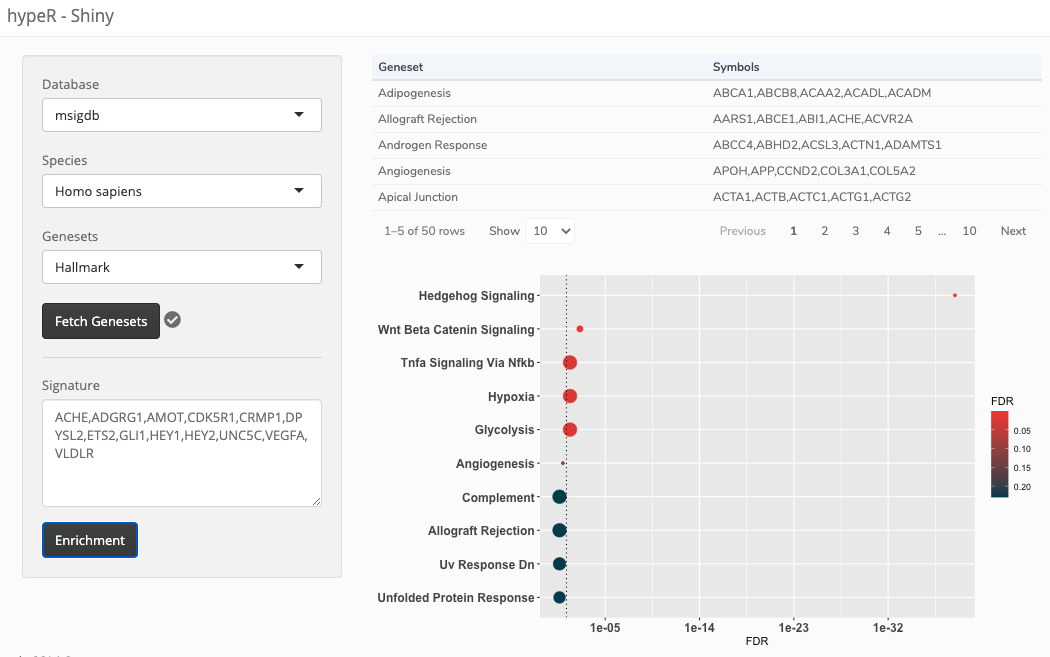
All hypeR methods output objects that are compatible with R Shiny and can be incorporated into a variety of applications (e.g. hyp_dots() returns a ggplot object). The most difficult challenge in incorporating geneset enrichment tasks in Shiny applications is the geneselect selection / fetching itself. To help with this, hypeR includes module functions that abstract away the geneset selection code.
What Are Shiny Modules?
Shiny modules are functions that generato Shiny UI / Server code. They are composable elements that can be used within and interface with existing applications. For more information, check out the latest documentation.
Example
For those familiar with Shiny, you will have accompanying modules for the ui and server sections of your application. These modules are hypeR::genesets_UI() and hypeR::genesets_Server() respectively.
UI
Here we create a simple interface where we use the genesets selection module. Through that module, users can select genesets available through hypeR and the application will load those genesets into a reactive variable (more on this later). We add additional code to take a user-defined gene signature and produce an enrichment plot which represents components specific to your Shiny application. This can be anything!
ui <- fluidPage( sidebarLayout( sidebarPanel( # Put your geneset selector module anywhere hypeR::genesets_UI("genesets"), # Add components specific to your application textAreaInput("signature", label="Signature", rows=5, placeholder="GENE1,GENE2,GENE3", resize="vertical"), actionButton("enrichment", "Enrichment") ), mainPanel( # Enrichment plot plotOutput("plot") ) ) )
Server
Now we need to create the server code that is responsible for all the backend work. Again, we provide an associated module function for the server code. The serve code returns a reactive variable containing the fetched genesets that were selected. Selection changes will update this variable and propogate to downstream functions. Importantly, this variable holding the genesets can be accessed by any part of your application now, enabling you to make applications completely customizable while still utilizing this feature. As an example, our custom function is producing a simple enrichment plot.
server <- function(input, output, session) { # Retrieve selected genesets as a reactive variable # Selection changes will update this variable and propogate to downstream functions genesets <- hypeR::genesets_Server("genesets") # Your custom downstream functions reactive_plot <- eventReactive(input$enrichment, { # Here are the fetched genesets gsets <- genesets() # Process the signature into a character vector signature <- input$signature %>% stringr::str_split(pattern=",", simplify=TRUE) %>% as.vector() # Run hypeR hyp <- hypeR::hypeR(signature, gsets, test="hypergeometric") p <- hypeR::hyp_dots(hyp, top=10, fdr=0.25) # These are just ggplot objects you could customize p + theme(axis.text=element_text(size=12, face="bold")) }) output$plot <- renderPlot({ reactive_plot() }) }
Session Info
R version 4.0.0 (2020-04-24)
Platform: x86_64-pc-linux-gnu (64-bit)
Running under: Ubuntu 18.04.4 LTS
Matrix products: default
BLAS/LAPACK: /usr/lib/x86_64-linux-gnu/libopenblasp-r0.2.20.so
locale:
[1] LC_CTYPE=en_US.UTF-8 LC_NUMERIC=C
[3] LC_TIME=en_US.UTF-8 LC_COLLATE=en_US.UTF-8
[5] LC_MONETARY=en_US.UTF-8 LC_MESSAGES=C
[7] LC_PAPER=en_US.UTF-8 LC_NAME=C
[9] LC_ADDRESS=C LC_TELEPHONE=C
[11] LC_MEASUREMENT=en_US.UTF-8 LC_IDENTIFICATION=C
attached base packages:
[1] stats graphics grDevices utils datasets methods base
other attached packages:
[1] reactable_0.2.0 forcats_0.5.0 stringr_1.4.0
[4] dplyr_1.0.0 purrr_0.3.4 readr_1.3.1
[7] tidyr_1.1.0 tibble_3.0.3 ggplot2_3.3.2
[10] tidyverse_1.3.0 hypeR_1.5.1 hyperworkshop_1.5.0
loaded via a namespace (and not attached):
[1] fs_1.4.1 usethis_1.6.1 lubridate_1.7.9 devtools_2.3.1
[5] webshot_0.5.2 httr_1.4.2 rprojroot_1.3-2 tools_4.0.0
[9] backports_1.1.8 R6_2.4.1 DBI_1.1.0 colorspace_1.4-1
[13] withr_2.2.0 tidyselect_1.1.0 prettyunits_1.1.1 processx_3.4.3
[17] curl_4.3 compiler_4.0.0 cli_2.0.2 rvest_0.3.6
[21] xml2_1.3.2 desc_1.2.0 labeling_0.3 scales_1.1.1
[25] callr_3.4.3 pkgdown_1.5.1 digest_0.6.25 rmarkdown_2.3
[29] pkgconfig_2.0.3 htmltools_0.5.0 sessioninfo_1.1.1 dbplyr_1.4.4
[33] fastmap_1.0.1 readxl_1.3.1 htmlwidgets_1.5.1 rlang_0.4.7
[37] rstudioapi_0.11 shiny_1.5.0 visNetwork_2.0.9 farver_2.0.3
[41] generics_0.0.2 jsonlite_1.7.0 crosstalk_1.1.0.1 zip_2.0.4
[45] magrittr_1.5 kableExtra_1.1.0 Rcpp_1.0.5 munsell_0.5.0
[49] fansi_0.4.1 lifecycle_0.2.0 stringi_1.4.6 yaml_2.2.1
[53] MASS_7.3-51.6 pkgbuild_1.1.0 plyr_1.8.6 blob_1.2.1
[57] grid_4.0.0 promises_1.1.1 crayon_1.3.4 haven_2.3.1
[61] msigdbr_7.1.1 hms_0.5.3 knitr_1.29 ps_1.3.3
[65] pillar_1.4.6 igraph_1.2.5 reshape2_1.4.4 pkgload_1.1.0
[69] reprex_0.3.0 glue_1.4.1 evaluate_0.14 modelr_0.1.8
[73] remotes_2.2.0 vctrs_0.3.2 tweenr_1.0.1 httpuv_1.5.4
[77] cellranger_1.1.0 testthat_2.3.2 gtable_0.3.0 polyclip_1.10-0
[81] reactR_0.4.3 assertthat_0.2.1 xfun_0.16 ggforce_0.3.2
[85] openxlsx_4.1.5 mime_0.9 xtable_1.8-4 broom_0.7.0
[89] later_1.1.0.1 viridisLite_0.3.0 memoise_1.1.0 ellipsis_0.3.1